CHROMOSOME ANOMALIES
Chromosome anomalies ( i.e., abnormalities, aberrations) may be of two general types :
a. Numerical
b. Structural
5% of recognised pregnancies are chromosomally abnormal and 0.5% of newborns have a chromosome abnormality.
NUMERICAL ALTERATIONS
POLYPLOIDY
Triploidy has 69 chromosomes with XXX, XXY or XYY. It may result from a failure of meiosis in a germ cell or dispermy. Tetraploidy or 92 chromosomes results from a failure of the first cleavage division after fertilization which results in a doubling of the chromosomes.
In both triploidy and tetraploidy, live births are rare. Liveborn triploidy infants survive briefly with clinical features like a relatively large head, congenital heart lesions and syndactyly. The placenta is small with hyaditiform changes. The features in a rare ongoing tetraploidy pregnancy are marked growth retardation, microcephaly and multiple malformations.
TRISOMY
It is the presence of three copies of a chromosome instead of two. The commonest cause is nondisjunction in meiosis I or II in either sex or in an early mitotic division of the zygote.Most trisomic embryos are lost in early pregnancy. Viability depends on the size of the imbalance and the chromosome involved. Usually only trisomies of 13, 18 and 21 survive to term and are seen in the liveborn population. Trisomy of the sex chromosomes such as XXY and XXX have less severe effects on development and do survive.
Trisomy 21
The infant is hypotonic and has a characteristic face with a flat nasal bridge, epicanthic folds, hypertelorism, Brushfield spots in the iris, protruding rough, ridged tongue, small low set ears and a flat occiput. The skin is dry and rough. The hands are stubby, with simian creases and clinodactyly and aplasia or hypoplasia of the middle phalanx of the little finger. A wide 'scandle gap' between 1st and 2nd toes is also seen. Cardiac lesions like ASD, VSD and an atrioventricular canal may be seen. Duodenal atresia may occur. As growth proceeds, milestones are delayed and short stature becomes a common feature. Mental retardation becomes evident ranging from moderate to severe. Hearing loss, autoimmune disorders, and the greater possibility of developing leukemia and Alzheimer's disease in old age are some of the other features.
Trisomy 13
Most severe cases abort spontaneosuly in the first trimester. Newborns show growth retardation and midline abnormalities like midline scalp defects, brain lesions like holoproscencephaly, and facial defects like central or unilateral clefts, hyopotelorism, microphthalmia or anophthalmia. Polydactyly, cardiac defects and omphalocoele along with severe mental retardation are some of the other features.
Trisomy 18
The newborns are severely handicapped with small facies, a prominent occiput, small ears, overlapping fingers and rocker-bottom feet. Cardiac as well as other internal malformations are usually seen.
Klinefelter syndrome (47, XXY)
In a young boy there will be a mild delay in development and immaturity. An older boy will have small, soft testes, while an adult male will have a eunuchoid habitus, gynaecomastia and poor musculature. Due to progressive hyalinization and fibrosis of the seminiferous tubules, there is infertility due to azoospermia or oligospermia and associated inadequate testosterone production.
47,XYY syndrome
The affected males are of tall stature with mild to severe social problems.Aggressive behaviour may be seen. Fertility is usually normal.
47,XXX syndrome
Apart from being taller than average, affected girls are physically normal. The mean IQ is lower than in controls and majority have educational problems. Gonadal functions are usually normal but oligomenorrhoea or premature ovarian failure may occur.
Nondysjunction or failure of chromosomes to segregate at mitosis or any of the two meiotic divisions produce trisomies. It is more common in oogenesis than in spermatogenesis and in meiosis I than in meisois II. The incidence of trisomy 13,18 and 21 births increase with maternal age and show a sharp rise over 35 years. Of the sex chromosome trisomies, only 47, XXY is associated with advanced maternal age.
MONOSOMY
It is the presence of only one member of a chromosome pair. Autosomal monosomies are lethal but the monosomy for the X chromosome, 45,X (Turner syndrome) is compatible with life, although it is common in aborted embryos.
Turner syndrome 45,X
Infants in this category may have neck webbing, shield shaped chest, coarctation of the aorta and lymphoedema of hands and feet. In childhood they present with short stature or a cardiac murmur. Teenagers may present with primary or secondary amenorrhoea, lack of secondary sexual characters due to the streaky ovaries , broad shield shaped chests. widely spaced nipples, cubitus valgus and spatial perceptual abnormalities. Mentally they are normal. Adults may present with infertility.
Nondisjunction or chromosome lag contributes to the monosomy. A lag of the Y chromosome at meiosis of spermatogenesis is thought to be a common cause of 45, X Turner syndrome. Mosaicism with a 45,X and a 46,XX cell line or a 45,X with a 46,XY cell line have been reported.
MOSAICISM
It is the presence of two or more cell lines with different karyotypes in a person. Usually a normal cell line exists with another normal cell line of the opposite sex or with an abnormal cell line. For example : 46,XX / 46,XY, or a 46, XX / 47,XX, + 21. The abnormal cell line may be confined to a particular tissue if the aberration took place in late embryonic or foetal development or multiple tissues if the changes took place in very early development. The clinical features will depend on the chromosome involved and the proportion of abnormal to normal cells.
STRUCTURAL ALTERATIONS
These result from breaks or breaks followed by fusion of chromosome segments in various ways. Fig. 22. As a result, acentric fragments and dicentric chromosomes arise which are unstable at cell division. These are lost and result in cell death. However, stable alterations are also seen.
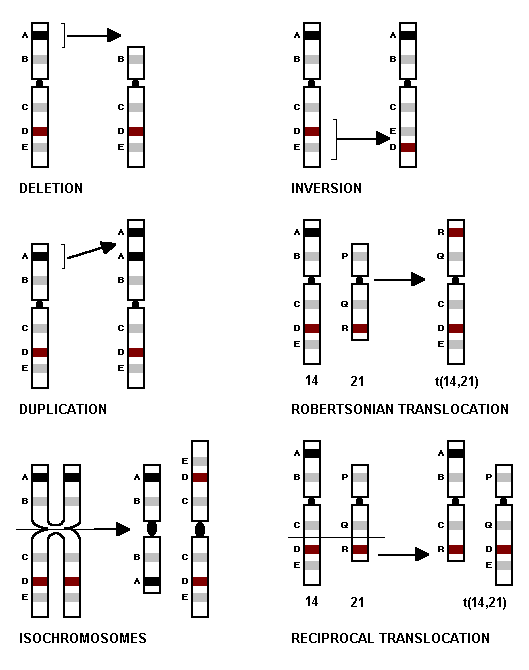
Fig 22
DELETION
These lead to a loss of chromatin. In terminal deletions the break is at the end of an arm. Cri du chat syndrome has the tip of the short arm of chromosome No. 5 deleted (5p-). These children have a round face, are mentally retarded and have a high pitched cat like cry. In interstitial deletions two breaks occur within the chromosome arms, the broken segment is lost and the ends unite to form a short arm. In ring chromosome disorders, two breaks occur at the tips of both arms of the chromosome, acentric pieces are lost from the ends, and the two broken arms curl towards each other and fuse to form a ring.
In microdeletion syndromes, very small deletions take place and are detected only by special high resolution banding. An example is Prader - Willi syndrome involving the long arm of chromosome No. 15.
DUPLICATION
They represent a gain of chromosome material by doubling, in a particular region.
ISOCHROMOSOME
These are formed when a chromosome with two chromatids splits at right angles to the normal lengthwise division seen at cell division. The resulting chromosomes will have either both short arms or both long arms. The nett loss will be an entire long arm or an entire short arm. Twenty percent of Turner syndrome result from having one normal X and another X which is an isochromosome for the long arm, essentially lacking a short arm.
INVERSION
In these the chromosomes break at two points and the intervening broken segment turns round 180 degrees to reverse the order of chromatin. If the break points are on the same arm it is called a paracentric inversion and if it is on either side of the centromere, including it in the broken segment, it is referred to as a pericentric inversion.
ROBERTSONIAN TRANSLOCATION
This results from a fusion of whole arms of acrocentric chromosomes. Fig. 22. The breakpoints are at or near the centromeres of both chromosomes. The fused long arm chromosome survives while the fused short arm chromosome is lost. This loss produces no effect as the short arms of acrocentrics contain genetically inert material or RNA genes.
A common example of such a translocation is seen in Down syndrome. It involves the long arms of chromosomes 14 and 21 - t (14q21q). The Down patient will have 3 of chromosome No 21 of which one is translocated. The balanced translocation carrier however will have only 2 copies of chromosome No 21 one of which will be translocated. Basically the chromosome complement will be normal, but with one translocated.
RECIPROCAL TRANSLOCATION
This results from breakage and exchange of segments between chromosomes. Fig. 22. The points of exchange may be anywhere along the chromosome. The reciprocal translocations show exchange of segments with no loss of chromosome material.
OTHER ANOMALIES
CHROMOSOME FRAGILE SITES
A site at the tip of the long arm of chromosome X shows increased fragility when blood is cultured in low folic acid culture media. The condition is called the Fragile X mental retardation syndrome. The affected boys show moderate educational subnormality and a speech delay or disorder. They have closely set eyes with large ears, a triangular face and prominent mandibles. The testis shows macroorchidism. Females are less frequent and those with one fragile X are mentally normal.
CHROMISOME BREAKAGE SYNDROMES
Here multiple visible breaks are seen due to faulty DNA repair and synthesis. Radiation or chemicals may be involved. Fanconi Anaemia and Ataxia Telangiectasia show such multiple fragility. Blooms syndrome is another breakage syndrome which exhibits increased sister chromatid exchanges when specially cultured. Here sister chromatids break and exchange segments, all within a single chromosome. All three conditions are autosomal recessive.